
O que é o Teste da Bochechinha?
O Teste da Bochechinha é uma triagem neonatal que analisa mais de 340 doenças genéticas raras que se manifestam ainda na infância e podem ser tratadas ou atenuadas quando diagnosticadas precocemente.
O objetivo desse teste é complementar o teste do pezinho expandindo o leque de doenças analisadas, garantindo segurança para a família e mais saúde ao bebê.
Conheça os Benefícios do Teste da Bochechinha
Ele é um teste genético complementar aos exames básicos e detecta de forma precoce e ainda na primeira infância condições raras e muitas vezes assintomáticas que podem ser tratadas com muita eficiência quando descobertas cedo.

É coletado com um swab (cotonete) a saliva do bebê no lado interno da bochecha, sem incômodos ou dores. No Laborali o teste pode ser realizado no conforto da sua casa.
Ao rastrear doenças permite que o tratamento com especialista se inicie imediatamente, melhorando a qualidade de vida do bebê.
É coletado com um swab (cotonete) a saliva do bebê no lado interno da bochecha, sem incômodos ou dores. No Laborali o teste pode ser realizado no conforto da sua casa.
A análise dos genes permite identificar mais de 340 doenças genéticas que vão desde as neurológicas até as endócrinas.
Quais doenças o Teste da Bochechinha detecta?
O Teste da Bochechinha permite uma análise minuciosa das seguintes doenças e genes:
- Acidose Tubular Renal Distal – ATP6V0A4
- Acidose Tubular Renal Distal e Surdez Neurosensorial Progressiva – ATP6V1B1
- Diabetes Insipidus Nefrogênico – AQP2 e AVPR2
- Síndrome de Bartter – BSND, CLCNKA, CLCNKB, KCNJ1 e SLC12A1
- Síndrome de Fechtner – MYH9
- Deficiência Combinada de Hormônios Hipofisários – LHX3, LHX4, OTX2, POU1F1, PROP1 e SOX3
- Deficiência de Corticosterona Metiloxidase – CYP11B2
- Deficiência de Diidrolipoamida Desidrogenase – DLD
- Deficiência de Glicocorticóide – MC2R, MRAP e NNT
- Deficiência de Transportador de Monocarboxilato 1 e Hipoglicemia Hiperinsulinêmica – SLC16A1
- Deficiência de TRH – TRH
- Diabetes Neonatal – INS
- Diabetes Neonatal e Hipotireoidismo Congênito – GLIS3
- Displasia Septo-óptica – HESX1
- Hiperparatireoidismo Neonatal Grave – CASR
- Hiperplasia Adrenal Congênita – CYP11B1, CYP17A1 e HSD3B2
- Hiperplasia Adrenal Congênita Lipóide – STAR
- Hipoglicemia Hiperinsulinêmica – GLUD1 e INSR
- Hipoglicemia Hiperinsulinêmica e Diabetes Neonatal – ABCC8, GCK e KCNJ11
- Hipoplasia Adrenal Congênita – NR0B1
- Hipotireoidismo Central Congênito – IRS4
- Hipotireoidismo Central e Surdez – TBL1X
- Hipotireoidismo Congênito – IGSF1, NKX2-5, PAX8, THRA, TRHR, TSHB e TSHR
- Pseudohipoaldosteronismo – SCNN1A, SCNN1B e SCNN1G
- Raquitismo Deficiente de Hidroxilação de Vitamina D – CYP27B1 e CYP2R1
- Raquitismo Dependente de Vitamina D – VDR
- Raquitismo Hipofosfatêmico – PHEX
- Osteogênese Imperfeita – COL1A1 e COL1A2
- Afibrinogenemia Congênita – FGA
- Anemia Falciforme, Talassemia e Outras Hemoglobinopatias – HBB
- Deficiência Combinada de Fatores de Coagulação Dependentes de Vitamina K – GGCX e VKORC1
- Hemofilia A* (*Não inclui inversão intrônica) – F8
- Hemofilia B – F9
- Púrpura
- Trombocitopênica Trombótica Congênita – ADAMTS13
- Trombocitopenia Amegacariocítica Congênita – MPL
- Colestase e Surdez – USP53
- Colestase Intra-hepática Familial Progressiva – ABCB4, ATP8B1, TJP2 e ABCB11
- Deficiência Congênita de Lactase – LCT
Diarréia Congênita – DGAT1, NEUROG3 e SLC26A3 - Intolerância a Fructose – ALDOB
- Malabsorção de Glicose e Galactose – SLC5A1
- Síndrome de Crigler-Najjar – UGT1A1
- Deficiência de Linfócitos T – ZAP70
- Deficiência de Linfócitos T, Alopecia Congênita e Distrofia Ungueal – FOXN1
- Deficiência de Mieloperoxidase – MPO
- Deficiência de Purina Nucleosídeo Fosforilase – PNP
- Desregulação Imunológica, Poliendocrinopatia e Enteropatia (IPEX) – FOXP3
- Disgenesia Reticular – AK2
- Doença Granulomatosa Crônica (CGD) – CYBA, CYBB, CYBC1, NCF2 e NCF4
- Imunodeficiência de Células T – CD247, CORO1A e ORAI1
- Imunodeficiência e Hiper-IgM – AICDA, CD40, CD40LG e UNG
- Imunodeficiência, Defeito do Magnésio, Infecção por Epstein-Barr e Neoplasia – MAGT1
- Infecções Piogênicas Recorrentes – MYD88
- Linfedema Primário, Mielodisplasia, Imunodeficiência e Leucemia Mielóide Aguda – GATA2
- Linfohistiocitose Hemofagocítica – PRF1, STX11, STXBP2 e UNC13D
- Neutropenia Congênita – CXCR2
- Neutropenia Congênita Grave – ELANE, G6PC3, GFI1, HAX1, JAGN1 e VPS45
- Síndrome de Falência da Medula Óssea – DNAJC21
- Síndrome de Shwachman-Diamond – EFL1, SBDS e SRP54
- Síndrome de Wiskott-Aldrich – WAS e WIPF1
- Síndrome do Linfócito Desnudo – CIITA, RFX5, RFXANK, RFXAP, TAP1, TAP2 e TAPBP
- Síndrome Linfoproliferativa – SH2D1A e XIAP
Síndrome WHIM – CXCR4 - Susceptibilidade a Infecções por Micobactérias – IFNGR2, IFNGR1, IL12B, IL12RB1, IRF8 e RORC
- Susceptibilidade a Infecções por Micobactérias e Virais – STAT1
- Atrofia Muscular Espinhal – SMN1
- Crises Encefalomiopáticas Metabólicas Recorrentes, Rabdomiólise, Arritmias Cardíacas e Neurodegeneração – TANGO2
- Distrofia Muscular de Duchenne – DMD
Hiperecplexia 1 – GLRA1 - Hiperecplexia 2 – GLRB
- Hiperecplexia 3 – SLC6A5
- Miastenia congênita associada a deficiência do receptor de acetilcolina – RAPSN
- Miopatia e Metabolismo Anormal de Lipídios (Deficiência de Desidrogenases Múltiplas) – FLAD1
- Neuropatia e Atrofia Óptica – PDXK
- Neuropatia por Sorbitol – SORD
- Fibrose Cística – CFTR
- Acidose Tubular Renal Distal – ATP6V0A4
- Acidose Tubular Renal Distal e Surdez Neurosensorial Progressiva – ATP6V1B1
- Diabetes Insipidus Nefrogênico – AQP2 e AVPR2
- Síndrome de Bartter – BSND, CLCNKA, CLCNKB, KCNJ1 e SLC12A1
- Síndrome de Fechtner – MYH9
- Retinoblastoma – RB1
- Acidúria Alfa-Metilacetoacética – ACAT1
- Acidúria Argininosuccínica – ASL
- Acidúria Glutárica (Acidemia Glutárica) – GCDH
- Acidúria Isovalérica (Acidemia Isovalérica) – IVD
- Acidúria Metilmalônica – CD320, MMAA, MMAB e MMUT
- Acidúria Metilmalônica e Homocistinúria – ABCD4, LMBRD1, MMACHC e MMADHC
- Acidúria Propiônica (Acidemia Propiônica) – PCCA e PCCB
- Adrenoleucodistrofia – ABCD1
- Alfa-manosidose – MAN2B1
- Argininemia – ARG1
- Cardiomiopatia e Degeneração de Retina Progressiva – SLC6A6
- Cistinose Nefropática – CTNS
- Cistinúria – SLC7A9
- Citrulinemia – ASS1 e SLC25A13
- Defeito de Síntese de Ácidos Biliares – AKR1D1, AMACR, CYP7A1, CYP7B1, HSD3B7 e SLC27A5
- Deficiência Cerebral de Creatina – GAMT e GATM
- Deficiência de Fosfoenolpiruvato Carboxiquinase – PCK1
- Deficiência de 3-Hidroxi-3-Metilglutaril-CoA Sintase 2 – HMGCS2
- Deficiência de 3-Hidroxi-3-Metilglutaril-CoA Liase – HMGCL
- Deficiência de 3-Hidroxi-acil-CoA Desidrogenase – HADH
- Deficiência de Ácido Ceto de Cadeia Ramificada Desidrogenase Kinase – BCKDK
- Deficiência de Acil-CoA Desidrogenase de Cadeia Média – ACADM
- Deficiência de Acil-CoA Desidrogenase de Cadeia Muito Longa – ACADVL
- Deficiência de Acil-CoA Desidrogenase Múltipla – ETFA, ETFB e ETFDH
- Deficiência de Biotinidase – BTD
- Deficiência de Carbamoil Fosfato Sintetase I – CPS1
- Deficiência de Carnitina Palmitoiltransferase I – CPT1A e II Infantil – CPT2
- Deficiência de Carnitina-Acilcarnitina Translocase – SLC25A20
- Deficiência de Fosfoglicerato Desidrogenase – PHGDH
- Deficiência de Fosfoserina Aminotransferase – PSAT1
- Deficiência de Fosfoserina Fosfatase – PSPH
- Deficiência de Frutose-1,6-Bisfosfatase – FBP1
- Deficiência de Galactoquinase (Galactosemia) – GALK1
- Deficiência de Galactose Epimerase (Galactosemia) – GALE
- Deficiência de Holocarboxilase Sintetase – HLCS
- Deficiência de L-Amino Ácido Aromático Descarboxilase – DDC
- Deficiência de Lipase Ácida Lisossomal – LIPA
- Deficiência de N-Acetilglutamato Sintase – NAGS
- Deficiência de Ornitina Transcarbamilase – OTC
- Deficiência de Piridoxamina 5-Primo-Fosfato Oxidase – PNPO
- Deficiência de Proteína Trifuncional Mitocondrial – HADHA
- Deficiência de Proteína Trifuncional Mitocondrial – HADHB
- Deficiência de Succinil-CoA:3-Oxoácido-CoA Transferase – OXCT1
- Deficiência de Sucrase-Isomaltase – SI
- Deficiência de Timidina Quinase – TK2
- Deficiência Sistêmica Primária de Carnitina – SLC22A5
- Distonia Dopa-Responsiva – SPR
- Distúrbio Congênito da Glicosilação (CDG) – Tipo Ib – MPI, Tipo It – PGM1 e Tipo Iz – CAD
- Doença de Gaucher – GBA
- Doença de Segawa – TH
- Doença do Xarope de Bordo – BCKDHA, BCKDHB e DBT
- Encefalopatia Etilmalônica – ETHE1
- Encefalopatia Relacionada ao Transporte de Malato-Aspartato – GOT2
- Fenilcetonúria – PAH
- Galactosemia – GALT
- Glicogenose – Tipo 0A – GYS2, Tipo 0B – GYS1, Tipo IA – G6PC1, Tipo IB/IC – SLC37A4, Tipo III – AGL, Tipo IV – GBE1, Tipo IXA1 – PHKA2, Tipo IXB – PHKB, Tipo IXC – PHKG2, Tipo VI – PYGL e Tipo XII (Deficiência de Aldolase) – ALDOA
- Hipercolanemia – BAAT
- Hiperfenilalaninemia Deficiente de BH4 – GCH1, PCBD1, PTS e QDPR
- Hiperfenilalaninemia, Distonia e Deficiência Intelectual – DNAJC12
- Hipofosfatasia – ALPL
- Homocistinúria – MTHFR
- Homocistinúria e Anemia Megaloblástica – MTR e MTRR
- Intolerância à Proteína Lisinúrica – SLC7A7
- Leucodistrofia Metacromática – ARSA
- Lipofuscinose Ceróide Tipo 2 (CLN2) – TPP1
- Malabsorção de Folato Hereditária – SLC46A1
- Mucopolissacaridose – Tipo I (Síndrome de Hurler e/ou Scheie) – IDUA, Tipo II – IDS, Tipo IVA – GALNS, Tipo VI – ARSB e Tipo VII – GUSB
- Neurodegeneração de Manifestação na Infância Responsiva a Biotina – SLC5A6
- Porfiria Eritropoiética Congênita – UROS
- Quilomicronemia – APOA5, APOC2, GPIHBP1, LMF1 e LPL
- Síndrome de Anemia Megaloblástica Responsiva a Tiamina – SLC19A2
- Síndrome de Deficiência de GLUT1 – SLC2A1
- Síndrome de Hiperornitinemia-Hiperamonemia-Homocitrulinúria – SLC25A15
- Síndrome Fanconi-Bickel – SLC2A2
- Síndrome MIRAGE – SAMD9
- Sitosterolemia – ABCG5 e ABCG8
- Xantomatose Cérebro-tendínea – CYP27A1
- Surdez – GJB2 e GJB6
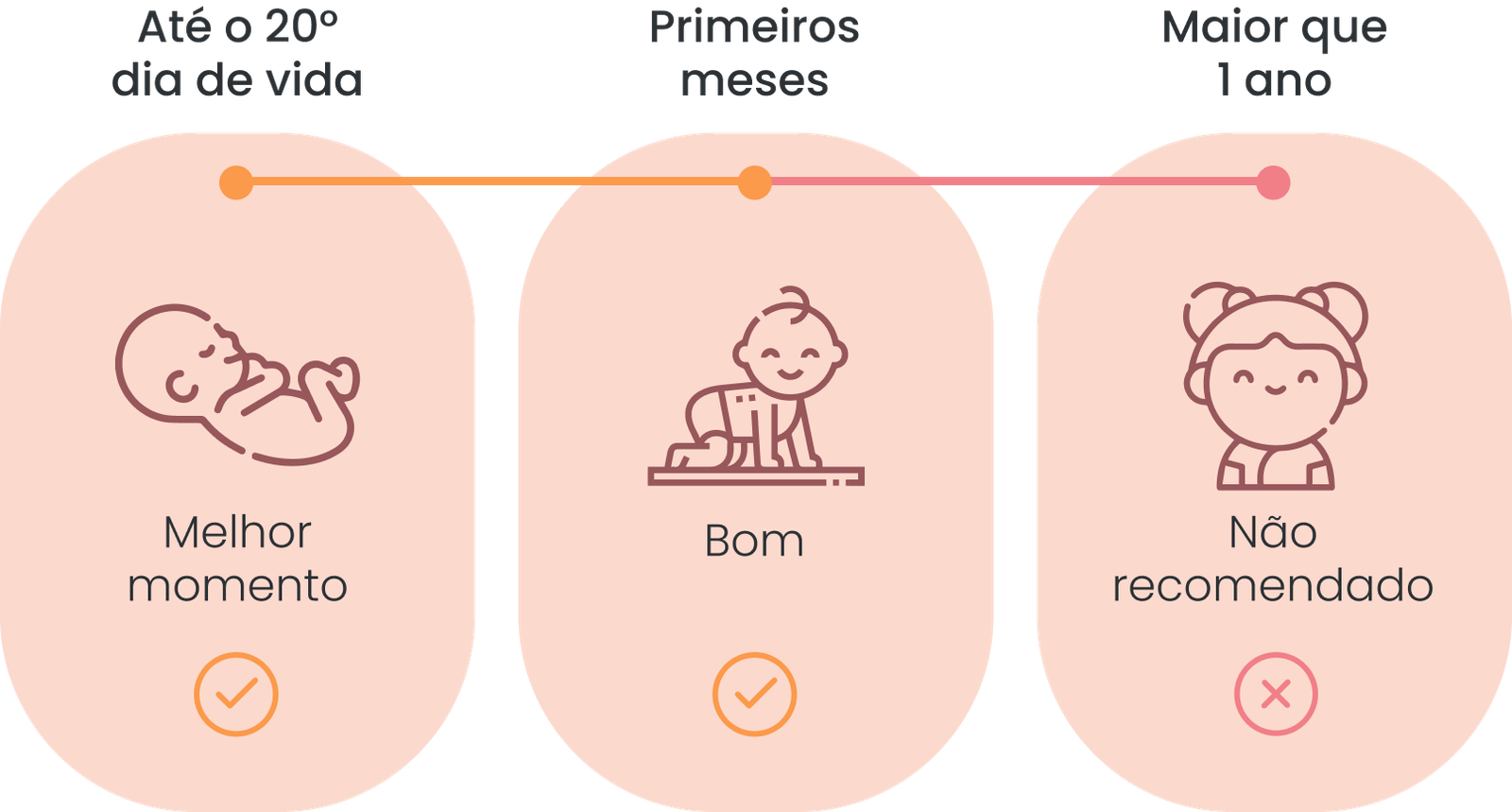
Até que idade o Teste da Bochechinha pode ser realizado?
O teste genético pode ser realizado a partir do 1º dia de vida do bebê para garantir um diagnóstico preciso e garantir tratamento precoce.
O bebê pode ser testado até completar um ano, após isso o procedimento não é recomendado.
Como o teste da bochechinha é realizado?

Coleta
A coleta é feita de forma rápida e indolor com um swab na mucosa bucal do bebê (na parte interna da bochechinha).

Análise
Após a coleta, o DNA é sequenciado e analisado em busca de alterações genéticas responsáveis por causar doenças.

Resultado
O DNA é analisado e você recebe o resultado em cerca de 28 dias.
Para realizar o teste, você pode se dirigir ao Laborali ou solicitar um atendimento domiciliar.
Observações e Curiosidades
Uma hora antes da coleta, a criança não deverá ingerir alimentos e líquidos ou inserir qualquer objeto na boca, como chupetas, mamadeiras ou mordedores.
O Teste da Bochechinha analisa cerca de 390 genes associados a condições que se iniciam na infância, incluindo muitas que podem ser tratadas imediatamente.
O Teste da Bochechinha não substitui o Teste do Pezinho, pois algumas doenças de início precoce podem ter causas que não são genéticas e por isso esse teste não detecta. Por isso, é muito importante realizar os dois testes!
Por que realizar o Teste da Bochechinha no Laborali?
Desde 1994 o Laboratório Laborali tem tradição em cuidar. Nosso compromisso é oferecer à comunidade uma vasta gama de exames com excelência e assertividade, bem como cuidar de todos para que sintam-se amparados.
Temos uma equipe preparada para atender pessoas de todas as idades com empatia, afeto e respeito.
Nossa equipe possui profissionais especializados e qualificados para bem atender os pequenos.
Ao atender crianças, usamos protocolos de comunicação não violenta, muita conversa e coleta de forma lúdica.
Dúvidas frequentes
Identifica precocemente diversas doenças graves que podem se manifestar na primeira infância, de forma assintomática. Essa avaliação rápida proporciona a possibilidade de tratamentos mais adequados a cada doença, evitando sequelas e danos à saúde e qualidade de vida.
Muitas dessas condições genéticas não são identificadas em outros exames neonatais.
Não, os exames de triagem neonatal são complementares, pois algumas doenças que não são detectadas no Teste do Pezinho, são reveladas com o Teste da Bochechinha e vice-versa. Além disso, como utilizam diferentes metodologias para análise do material biológico, recomenda-se que ambos os exames sejam realizados para que o seu bebê tenha um diagnóstico preciso e os melhores cuidados possíveis.
Além do que a detecção de doenças através do Teste do Pezinho realizado na rede pública de saúde, garante tratamentos custeados pelo SUS.
Não. Ele é um exame com finalidade clínica que atua como uma complementação a triagem neonatal.
O resultado informará apenas baixo risco (ausência de variantes associadas a quadro clínico), risco intermediário ou alto risco. Não informará se o bebê é portador assintomático de variantes sem risco clínico ou de condições de início tardio.
O exame é voltado para crianças assintomáticas. Portanto, caso o bebê tenha sintomas, suspeita clínica de doenças genéticas e indicação médica para realizar um exame genético.
Não é necessário pedido médico, porém, é muito importante que o seu bebê seja acompanhado por um pediatra, para a interpretação dos resultados e tratamento adequado.
Caso seja identificado alto ou moderado risco para desenvolvimento de condições genéticas, recomendamos que consulte um especialista imediatamente.
O laudo é disponibilizado em até 28 dias úteis após a coleta do exame.
Agende seu exame!
Agora que você já sabe a importância do teste da bochechinha, fale com nossa equipe para agendar o exame do seu bebê!